Catecholamine-secreting tumors are rare neoplasms which originate from chromaffin cells of the adrenal medulla (pheochromocytomas) or the sympathetic ganglia (in this case they are called catecholamine-secreting paragangliomas). They occur at any age but are most common in the fourth to fifth decade and are equally common in men and woman. Most of these tumors are sporadic; however, in a non-negligible percentage of cases (approximately 40 percent), they can be part of familial disorders which have an autosomal dominant inheritance (von Hippel-Lindau syndrome, multiple endocrine neoplasia type 2 and, less commonly, neurofibromatosis type 1).
Only 50 percent of patients with pheochromocytoma shows symptoms, and when present, they are typically paroxysmal. The most common symptoms are hypertension (both sustained and paroxysmal), headache, generalized sweating, palpitations, tremor, pallor, dyspnea, generalized weakness, and panic attack-type symptoms. The presence of all classic triad’s symptoms (episodic headache, sweating, and tachycardia) is described in a low percentage of patients1,3-5. Many other less common symptoms may occur and they include abnormalities in carbohydrate metabolism (insulin resistance, impaired fasting glucose, apparent type 2 diabetes mellitus), papilledema, weight loss, orthostatic hypotension, psychiatric disorders, increased erythrocyte sedimentation rate, visual blurring.
Pheochromocytoma multisystem crisis is a rare potentially lethal condition characterized by hypertension or hypotension, hyperthermia (temperature > 40°C), mental status changes and other organ dysfunctions which are treated with emergency surgery.
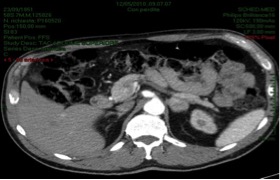
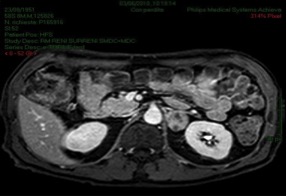
All symptoms are caused by tumoral hypersecretion of one or combinations of the following catecholamines: norepinephrine, epinephrine, and dopamine. For this reason, the diagnosis is based on biochemical confirmation of high values of these hormones (typically by measurements of urinary and plasma fractionated metanephrines and catecholamines)6 followed by identifying the tumor with imaging studies (CT, MRI, iodine-123 iobenguane scintigraphy and FDG-PET)1.
The treatment of adrenal pheochromocytoma is surgical and is preferable laparoscopic adrenalectomy by an experienced endocrine surgeon compared with open laparotomy7.
The preoperative preparation includes a pharmacological treatment with alpha-adrenergic blockade first and beta-adrenergic blockade at a later time8.