
- roberto ruffinengo
- Area giovani
Un caso di Rendu-Osler-Weber
- 4 - Settembre 2013
- ISSN 2532-1285
Abstract
A 26 years old man, with a history of cerebral arterio-venous malformation, comes to the Stroke Unit of the “Santa Maria della Misericordia” hospital for headache and partial seizures. An angioRM shows cerebral venous thrombosis. The patient receives heparin therapy but after a few days he develops anemia requiring transfusions due to recurrent epistaxis. It just seems an adverse effect of anticoagulant therapy but, together with some signs and symptoms and medical history, leads us to a single diagnosis.
Caso clinico
C.F., 26 anni, giunge presso la nostra Stroke Unit per la comparsa da alcuni giorni di cefalea a sede nucale, di moderata intensità, continua, non pulsante, ad esordio graduale, resistente ai comuni analgesici e senza altri sintomi associati. Non vengono riportati in anamnesi traumi cranici recenti né rialzo termico. Inoltre il paziente riferisce recente incremento della frequenza delle crisi epilettiche parziali sensitivo-motorie, caratterizzate da parestesie all’emivolto sinistro e al V dito della mano sinistra, deviazione dello sguardo verso sinistra e clonie dell’emivolto sinistro, per le quali già da alcuni anni era stato intrapreso trattamento anticonvulsivante con lamotrigina ed acido valproico. Tali manifestazioni neurologiche sono la conseguenza del trattamento chirurgico e radioterapico di una malformazione artero-venosa (MAV) cerebrale in sede frontale destra. In anamnesi risulta inoltre la presenza di
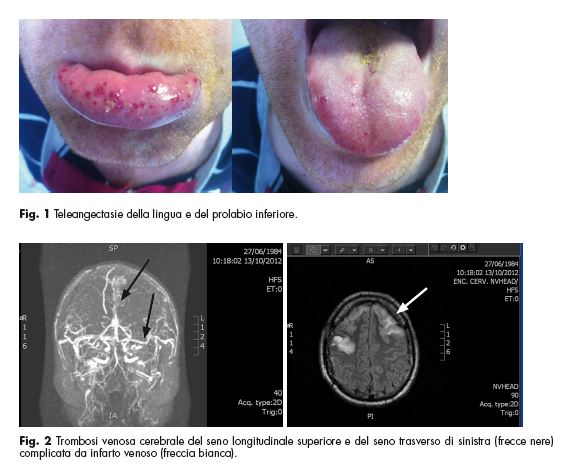
una MAV cervicale a livello di C2-C3, trattata con embolizzazioni multiple, con presenza di nidus angiomatoso residuo. All’anamnesi familiare risulta che anche il padre del ragazzo è portatore di MAV cervicali multiple. All’ingresso in reparto i parametri vitali risultano nella norma; l’esame obiettivo neurologico non mette in evidenza deficit focali di nuova insorgenza. All’esame obiettivo generale si evidenzia la presenza di teleangectasie della lingua e del prolabio inferiore (Fig.1). Per escludere una complicanza emorragica in paziente portatore di MAV si esegue una TC dell’encefalo in urgenza, risultata negativa per stravasi ematici acuti. Il dosaggio ematico dei farmaci antiepilettici è risultato in range, come nella norma sono risultati anche i restanti esami di laboratorio, eccezion fatta per una già nota anemia ferro-carenziale con valori di emoglobina pari a 11 g/dl. A distanza di 48 ore il paziente sviluppa uno stato di male epilettico. Pertanto viene intubato e trasferito al reparto di Rianimazione dove si esegue una RM dell’encefalo con sequenze angiografiche che mostra reperti compatibili con trombosi venosa cerebrale a carico del seno longitudinale superiore e del seno trasverso di sinistra complicata da infarto venoso (Fig.2). In considerazione dell’elevato rischio emorragico per la presenza di MAV cerebrali, inizialmente si decide di intraprendere terapia anticoagulante con eparina sodica in infusione continua (aPTT target 1.5), più
maneggevole e controllabile rispetto all’eparina a basso peso molecolare ed inoltre con un certo potenziale di reversibilità. Con tale terapia le condizioni cliniche generali e neurologiche gradualmente migliorano. Dopo circa 5 giorni il paziente viene nuovamente trasferito presso il nostro reparto per indagare la possibile secondarietà della trombosi venosa cerebrale. Le indagini eseguite in questo senso evidenziano solo una iperomocisteinemia pari a 34.4 mmol/l (v.n. <12), mentre lo screening per la trombofilia ereditaria, la ricerca degli anticorpi lupus
anticoagulant (LAC) e degli anticorpi antifosfolipidi (ACA) risulta negativo. Inoltre vengono eseguiti anche una radiografia del torace ed un’ecografia addominale, entrambi risultati nella norma. Durante la degenza si verificano numerosi episodi di epistassi, prevalentemente mattutina, non associati a rialzi pressori, che determinano il peggioramento dell’anemia ipocromica microcitica ferro-carenziale cronica fino a valori di Hb pari a 7.3 g/dl, che richiedono la trasfusione di tre unità di globuli rossi concentrati fino al raggiungimento
di valori stabili di Hb pari a 10.7 g/dl (Ht 36.5%, MCV 66 fl, MCH 19.5 pg, MCHC 29.3 g/dl, ferritina 2.4 ng/ml, sideremia 13 mcg/dl). I valori di piastrine e il quadro emostatico risultano nella norma. La ricerca del sangue occulto fecale risulta negativa in tre campioni. Si viene a conoscenza del fatto che il problema, inizialmente interpretato come complicanza della terapia eparinica, è in realtà presente già da molto tempo e risulta essere con tutta probabilità la causa dell’anemia ferro-carenziale. Viene pertanto effettuata una consulenza otorinolaringoiatrica, che evidenzia una mucosa nasale ricca di multiple lesioni simil-varicose con segni di sanguinamento recente. Lesioni ad aspetto reticolare sono inoltre presenti anche a livello della mucosa del labbro inferiore e della punta della lingua. Per lo spontaneo arresto dell’epistassi, non viene effettuato alcun tamponamento nasale.
In considerazione dei reperti clinico-laboratoristici, dell’anamnesi patologica remota e di quella personale e familiare si pone diagnosi di sindrome di Rendu-Osler-Weber (telangectasia emorragica ereditaria) secondo i criteri di Curaçao e si programma una consulenza genetica. Inoltre, alla luce dei dati della letteratura circa la possibile associazione di tale sindrome con un eccesso di fattore VIII si procede al dosaggio di tale fattore che risulta nella norma.
Il paziente viene dimesso in buone condizioni di salute. Vista la necessità di terapia anticoagulante, in considerazione della presenza di nido angiomatoso residuo a livello cervicale, si decide di proseguire la terapia con enoxaparina piuttosto che con anticoagulante orale con il programma di ridiscutere la terapia dopo 3 mesi previa esecuzione di RM encefalo di controllo.

Discussion
La sindrome di Rendu-Osler-Weber o teleangectasia emorragica ereditaria (Hereditary haemorrhagic telangiectasia: HHT) è una rara displasia sistemica fibro-vascolare, causata da una alterata angiogenesi che porta ad eccessiva neovascolarizzazione, fragilità della parete vascolare, dilatazione dei vasi e conseguente rottura.
Si tratta di una patologia congenita trasmessa con carattere autosomico dominante, sebbene in circa il 20% dei casi non vi sia storia familiare (1). La forma omozigote non è compatibile con la vita. I geni principalmente coinvolti nelle varie forme di HHT si trovano a livello del cromosoma 9q34.1, dove è codificata l’endoglina, corecettore di TGFb1 e TGFb3 (trasforming growth factor- b), che controlla la composizione delle adesioni focali e disciplina l’organizzazione di filamenti di actina, e a livello del cromosoma 12q31, dove è codificato ALK-1 (activina receptorlike chinasi 1), recettore TGFb1. Tali proteine si trovano espresse soprattutto a livello delle cellule endoteliali, dove mediano il legame con il TGF-b, una proteina coinvolta nei processi di rimodellamento della matrice cellulare (2). Si stima che l’incidenza di questa malattia nella popolazione generale sia di circa 1 caso su 5.000 abitanti (3-4), con una distribuzione omogenea per razza e genere (1). I pazienti affetti presentano teleangectasie multiple a livello della cute e delle mucose (naso, tratto gastro-enterico e cavo orale) e malformazione artero-venose a livello di diversi organi (cervello e midollo spinale, polmone, fegato, apparato gastro-enterico). Le manifestazioni cliniche sono molteplici ed hanno una diversa frequenza (5)(Tab.I). La più frequente è costituita da epistassi ricorrenti (6), che costituiscono la principale causa di anemia iposideremica. La presenza di anastomosi
artero-venose a livello polmonare, con conseguente shunt destro-sinistro, è responsabile del quadro clinico caratterizzato da scompenso ad alta portata con cianosi, tosse, dispnea, poliglobulia fino all’embolia paradossa (6;7;8). La radiografia del torace può mostrare una massa tipica con la dilatazione dell’arteria e della vena anche se l’esame che permette di visualizzare meglio le MAV è sicuramente l’angioTC. L’angiografia polmonare va utilizzata solo per la programmazione di un trattamento chirurgico o radiologico (9). La localizzazione a livello cerebrale o spinale può complicarsi più comunemente con emorragie spontanee ma anche con ischemia cerebrale ed ascessi cerebrali nel caso in cui il paziente presenti uno shunt polmonare destro-sinistro (10;11). A livello gastro-enterico le manifestazioni cliniche consistono in telangectasie simili a quelle della mucosa nasale ed orale, localizzate prevalentemente a livello dello stomaco e del duodeno, che causano stillicidio ematico cronico (6). Più raramente la malattia si manifesta con un’emorragia gastro-enterica acuta da rottura di MAV. Molto più raro è l’interessamento epatico, con MAV che possono stabilirsi tra vena e arteria epatica con scompenso cardiaco congestizio per meccanismo di shunt sinistro-destro, tra la vena porta e la vena epatica con il conseguente quadro di encefalopatia epatica ed infine tra l’arteria epatica e la vena porta causando ipertensione portale e sviluppo di varici esofagee (6;7). Ecografia addominale, Tc addome con mezzo di contrasto ed esami endoscopici possono essere usati a fini diagnostici. Da non dimenticare che questa malattia emorragica vascolare ha anche un potenziale profilo pro-trombotico legato alla possibile presenza di elevati livelli di fattore VIII, associata a un’incidenza pari al 6,5% di eventi trombo-embolici venosi (12).
La diagnosi si basa sui criteri di Curaçao, sviluppati tra il 1997 e il 1999 e recentemente validati (13). Si parla di “HHT certa” se sono soddisfatti almeno 3 criteri; “HHT probabile” se sono presenti 2 criteri e di “HHT improbabile” se è presente un solo criterio (Tab II). Per confermare la diagnosi, è attualmente disponibile un test genetico, che risulta di limitata utilità nel caso di HHT certa sulla base dei criteri clinici. Tuttavia lo screening genetico assume una certa rilevanza nei membri della famiglia in cui la diagnosi di HHT non può essere esclusa clinicamente, soprattutto nel caso in cui tale diagnosi possa determinare un rischio per le future generazioni. Per quanto riguarda i pazienti affetti da HHT o a rischio di HHT, questi devono essere sottoposti a esami strumentali (come per esempio gli esami endoscopici del tratto gastro-enterico superiore ed inferiore e l’angioTC polmonare) mirati ad identificare
la sede delle malformazioni viscerali, soprattutto se il paziente è di sesso femminile, a causa dell’elevato rischio di emorragie polmonari e cerebrali durante il primo trimestre di gravidanza. Appare invece dibattuta l’indicazione alla ricerca delle MAV cerebrali, poiché alcuni autori ritengono che le MAV cerebrali riscontrate in pazienti asintomatici
siano a minor rischio di rottura rispetto a quelle dei pazienti che presentano altri sintomi della malattia (14).
La terapia è di supporto e di prevenzione delle complicanze. Per quanto riguarda l’epistassi la terapia si basa essenzialmente sull’esecuzione di un tamponamento nasale anteriore o posteriore in base alla sede del sanguinamento con eventuale successiva cauterizzazione dei nidi ectasici più prominenti. Nel caso in cui l’epistassi non risponda a questi trattamenti e sia causa di importante anemia ferro-carenziale è possibile, con le moderne tecniche di imaging, procedere all’embolizzazione dell’arteria mascellare, etmoidale o sfenoidale o alla legatura
dell’arteria mascellare o palatina (6). Per quanto riguarda le lesioni viscerali le opzioni terapeutiche contemplano l’embolizzazione terapeutica, la radioterapia e la resezione chirurgica in caso di sanguinamenti recidivanti o importanti. Indiscussa appare la terapia trasfusionale di supporto e la terapia marziale. Per quanto riguarda il trattamento degli eventi tromboembolici venosi allo stato attuale la patologia non rappresenta una controindicazione alla terapia anticoagulante laddove ve ne sia necessità facendo come sempre una valutazione personalizzata sul singolo paziente del bilancio rischio-beneficio ed informandolo dei potenziali rischi emorragici (2).
